共振论(1)
在共轭二烯这一节的学习里,我们接触了所谓的“共振论”。这是传统有机化学家处理电子离域体系的一个常用方法。鉴于课本包括教学视频中 对它的介绍有些模糊,我们这里对共振论专门进行一些讨论。
基本原则
共振论从某个角度上说,是为传统结构理论打的一个“补丁”。这个补丁专门为处理共轭的电子离域体系而设计。
我们知道,在纸面上描绘有机分子的结构,我们通常会使用键线式、缩写式这样的所谓“经典Lewis结构式”,原子原子间通过共价键相互连接,一根共价键等同于一对电子。 而在Lewis经典结构式中,无论是单/双/叁键,成键都是局限于两个原子之间的。换而言之,成键原子固定在两个原子的范围之内,是所谓的“定域”的。 比如下图中1,3-丁二烯(80版系统命名)/丁-1,3-二烯(17版系统命名)的经典结构式, 从纸面上看,C1-C2与C3-C4之间都是双键,这两组原子间各有四个电子,而C2-C3间是单键,原子间有两个电子成键。
类似的,下图中的烯丙基碳正离子,纸面看C1-C2间四电子,C2-C3间两电子,正电荷完全分布在C3上。
但事实上,我们知道实际情况并非如此。如下图所示,像1,3-丁二烯,所有四个碳原子sp2杂化,四个p轨道侧面交叠可以形成四中心四电子大Π键, π电子可以在四个原子的大范围内自由流动,不再局限于两个原子之间,也即所谓的“离域”。此外,C2-C3间由于有p轨道的侧面交叠,实际也不仅仅是单键, 而是具有部分双键的性质。类似的,烯丙基碳正离子,三个碳原子也都是sp2杂化,形成三中心两电子大Π键,π电子也是离域的,C2-C3间同样具备部分双键的性质。 这些结构上的特征,在经典结构式中完全体现不出来。换句话说,使用电子定域的经典结构式,无法表征电子离域体系的实际结构。
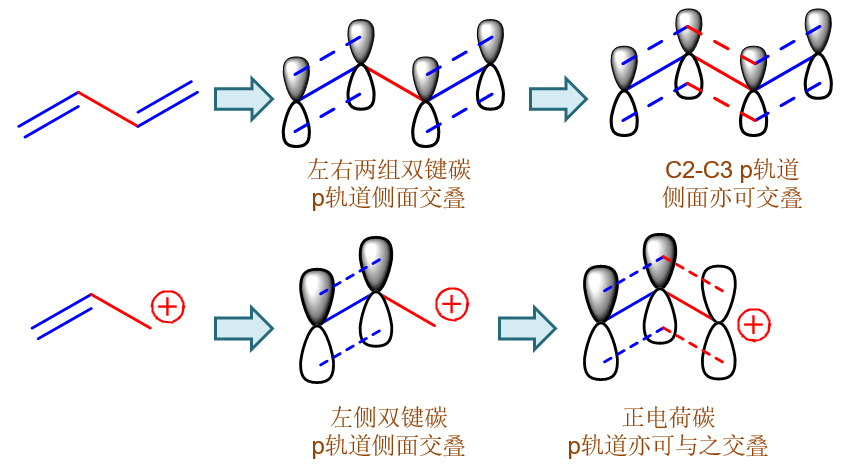
1,3-丁二烯与烯丙基碳正离子中均可形成电子离域的共轭体系
备注
关于共轭与电子离域的更多讨论,请见前一章的 重难点小结:诱导效应与共轭效应、碳正离子的稳定性。
基于这样的缺陷,“共振论”补丁出现了。共振论说白了就一个基本原则:
对于这种无法用单个经典结构式表达的电子离域体系,可以使用多个经典结构式(也即共振极限式)去表征。体系的实际结构相当于多个共振极限式的平均化。
非常直白也非常暴力的做法,一个经典结构式不行,就多上几个。
比如我们看到对1,3-丁二烯,可以用底下这么一大堆共振极限式来表达实际结构:
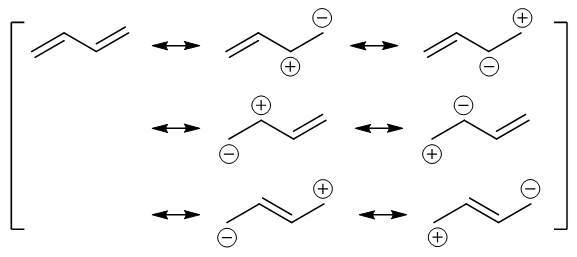
表达1,3-丁二烯实际结构式的一系列共振极限式
分子的实际结构被视作这些共振极限式的平均。而最后一行的两个极限式,纸面上C2-C3间是双键,平均之后,1,3-丁二烯的实际 结构里,C2-C3间也就带有了部分双键的成分。
共振论的大致原则就是如此。规则很简单,但这么一大堆极限式看起来挺晕,很多同学也不明白这些极限式是怎么得到、怎么画出来的。 接下来我们就专门来解释这个问题:如何从一个经典式得到一系列共振极限式。
共振极限式的画法
欲从经典结构式得到一系列共振极限式,其实说起来也就一个基本做法:人为地令π电子发生转移。
注意到我们当前用共振论处理的是电子离域的体系,π电子本身就是可以自由流动的,人为地令π电子转移,某种意义上说就是在模拟这种流动。 需要小心的是,这里令其流动的只是π电子,也就是经典结构式中π键及参与共轭的p轨道上的电子。反过来σ键上的电子不参与共轭,原本就是定域的, 在写共振极限式的时候切勿移动(换而言之,不要改变σ键)。
这么说还是比较抽象,我们不妨用一个具体的实例来说明。1,3-丁二烯的共振式比较复杂,我们先放到一边。咱们就以烯丙基碳正离子为例, 看看从下图中的经典结构式出发,如何画出它的共振极限式。
- 例:烯丙基碳正离子的共振式
-
首先,我们拎出这个结构中的π电子来。明显左侧C1-C2间纸面上的π键有一对π电子。此外从这个经典结构式上看,右侧C3碳正中心,有参与共轭的 p轨道,但这个p轨道是空的,没有电子。加到一起,这个结构总共也就是两个π电子。
接下来我们人为地令这一对π电子发生转移,从C1-C2间转移至C2-C3间:
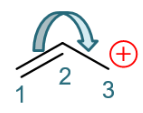
这样的转移,相当于C1-C2间π键一对电子移走了,双键会变成单键;而C2-C3间多了一对电子,单键变双键。正常共价键是每人出一个电子, 现在C3没出电子,平白生成了这么一根双键,相当于自己多接受了一个电子,正电荷刚好消失。于此同时C1-C2间旧的π键上两个电子现在全被C2拿去 成新键,自然C1相当于失去了一个电子,将带有正电荷。这两个电子转移后,我们将得到一个新的经典结构式:
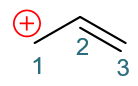
备注
这里我们还是用双钩弯箭头来标识一对电子的转移,但与之前在书写机理时的弯箭头含义略微有所不同。写机理的电子转移箭头,是 反应过程中实际发生的变化。而共振论这里,只是为了推衍新的共振极限式而作的人为的假定,并非实际的电子转移。写出的单个共振极限式 也并非分子或离子的实际结构。
好,这个新结构实际就是烯丙基碳正离子的又一个共振极限式。我们把原结构与新结构用共振箭头联系起来,如下图,这基本就是共振论 对烯丙基碳正离子结构的处理:
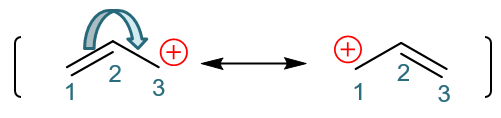
烯丙基碳正离子的一对共振极限式
烯丙基碳正离子的这个例子比较简单,使用同样的做法,我们可以来处理更复杂一些的1,3-丁二烯。
- 例:1,3-丁二烯的共振式
-
同样,我们首先还是先找出离域的π电子。明显C1-C2、C3-C4间的π键上,总共有两对离域的电子。老办法,我们人为令其发生转移。比如,先令 当前(a)式的C1-C2间一对电子转移到C1上,如下图所示。
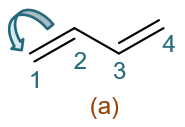
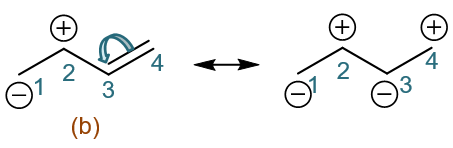
注意我们这是令一对电子从原子间转移到其中某一个原子上,相当于令π键发生异裂。这将导致C1带负电荷而C2带上正电荷。如是,我们将得到如下 电荷分离的一个共振极限式(b):
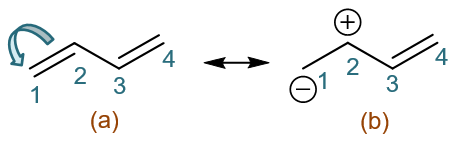
共振极限式(b)中,留意C2-C4这三个原子,我们看到这个区域其实就类似于之前的烯丙基碳正离子。同样的处理方式,我们可以人为令C3-C4间π键 上一对电子转移到C2-C3间,得到共振式(c):
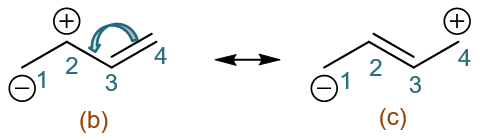
共振式(b)(c),可以说都是我们在(a)的基础上人为令电子转移到C1,使C2带正电荷一路衍生出来的。当然,我们也可以一开始就反过来,令C1-C2间的π电子反向转移, 全部转移到C2上,这么一来C2将带负电荷而C1带正电荷,又生成共振式(d):
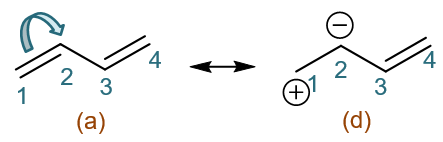
在共振式(d)中,C2-C4不再是烯丙基碳正离子的结构了,而是相反,接近于烯丙基碳负离子。而C2带负电荷,又位于双键旁侧,实际上它的杂化形态也是 sp2,一样能参与共轭。
在(d)的基础上我们继续变换。现在C2带负电荷,相当于其未参与杂化的p轨道上有一对电子(想想自由基碳p轨道1个电子,碳正0个,碳负自然是2个)。 我们人为令这对电子转移到C2-C3之间,使得这两个原子间由单键变成双键。于此同时,C3为了维持4价,需要断开一根旧键。σ键在写共振式的时候不能 发生变化,我们这儿也只能拆开C3-C4间的π键,令这对电子全部转移至C4,也即C4多接受一个电子,带上负电荷。这么一来我们又得到新共振式(e):
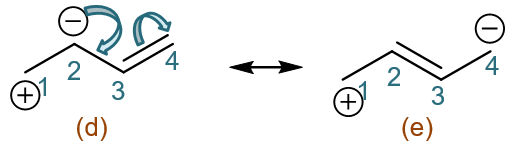
好,到目前为止我们在(a)的基础上首先拆开的都是C1-C2间的π键。类似的,我们也可以一上来就拆开C3-C4间的π键。比如先令一对电子都转移至C4, 同样可以衍生出共振式(f)与(g):
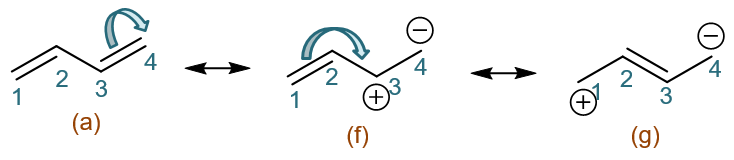
反过来令C3-C4间一对电子都转移至C3,又可以衍生出共振式(h)(i):
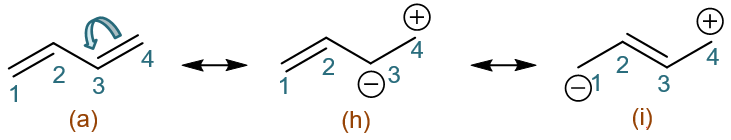
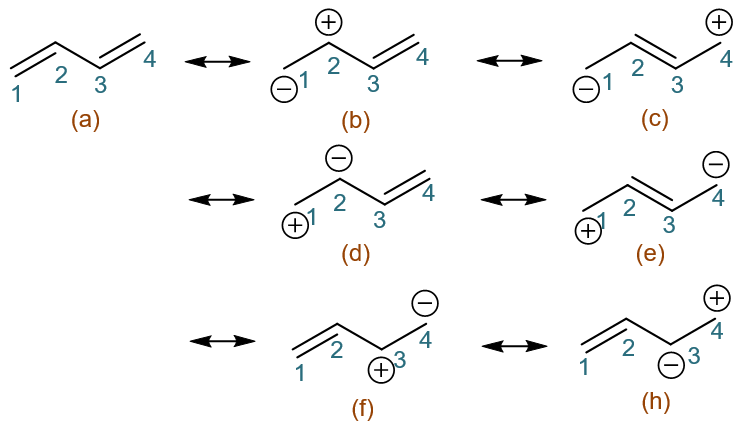
到这儿看起来我们一共得到了从(a)至(i)共九个共振极限式,但注意其中明显有几对重复的,如(c)与(i)、(e)与(g)。剔除重复,最终, 我们得到了1,3-丁二烯的共计七个比较重要的共振极限式:
备注
在这个例子中我们人为地将起始结构中的一根双键拆成单键,得到正负离子对。而1,3-丁二烯中经典结构式有两根双键,有些同学会有疑问,能否把 第二根双键也拆开,形成如下图中右侧的新共振式:
包括之前的烯丙基碳正离子,似乎也能拆开双键:
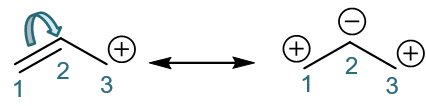
这样得到的新共振式行不行呢?答案其实是肯定的,但上面两个新共振式我们一般不用写出来,在讨论结构的时候也不必考虑。具体原因在 重难点小结:共振论(2)中会进行解释。
上面的例子里我们基本都是令一对电子发生转移,而后一路衍生。但实际上写共振式时,也可以令单电子转移。如下例中烯丙基自由基的共振式。
- 例:烯丙基自由基的共振式
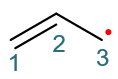
这个自由基的结构与烯丙基碳正离子相似,C3也是sp2杂化,只不过p轨道上不是空的,而是有一个电子。于是这个结构中, 参与共轭可以离域的π电子总共是三个:C1-C2间π键上一对,外加C3的p轨道上一个。
我们还是令电子发生转移。注意到当前C3上有单个的一个电子,我们不妨先令这未成键的一个电子转移到C2-C3之间。当然,光这单个电子 没法儿形成共价键,咱们还得再找个电子与它配对。当前其它碳原子上其实都没有多余的电子了,我们只能拆东墙补西墙,比如把C1-C2间 的π键断开,这么一来可以解放出两个电子。当然,前面咱们只需要一个,我们就令C1-C2间π键的一个电子转移到C2-C3间,刚好配对形成 一根新的π键。剩下一个电子不需要了,它只能留给C1,于是C1带上一个未成键电子,相当于自由基中心从C3转移到了C1上。整个过程大致 如下图所示:
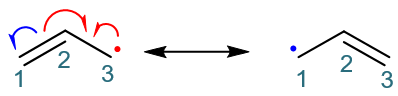
备注
在上面的过程中,发生的都是单电子的转移,因此这里使用的都是单钩箭头。
最终,烯丙基自由基的一对主要的共振极限式如下图所示:
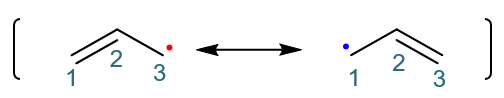
写共振式,大致就是这么一个过程,其实并不复杂。
备注
注意,我们书写共振式转移电子时,从不涉及σ键。而σ键保留,意味着分子/离子中原子的拓扑连接情况在所有共振式中都相同。 如以下的一对结构,σ键情况不同(C-H键连接情况发生了改变),绝不会是一对共振极限式。
