醛酮的亲核加成反应
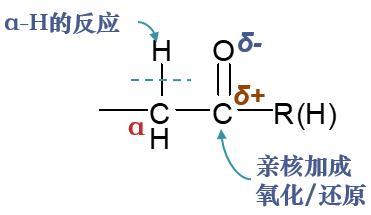
在基础有机里我们学习到的羰基化合物的化学反应中,比较重要的基本就三类:发生在羰基上的亲核加成反应与氧化/还原反应,以及由羰基旁侧活性α-H引发的相关反应。 这三大类别中,具体反应数量最多的无疑是亲核加成。
历程
万变不离其宗,醛酮亲核加成反应的数量虽多,但其历程却是大致相同的,基本过程如下图所示:
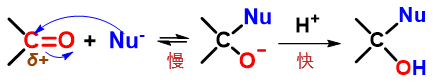
醛酮亲核加成典型历程A:亲核试剂直接进攻羰基碳
羰基中C=O双键明显是一根极性共价键,氧原子电负性高于碳,因而电子云会向氧原子富集,相应地碳原子上会缺电子,带有部分正电荷。 而这个缺电子碳为无疑会吸引亲核试剂(往往是负离子)向其靠拢,进而发动亲核进攻,形成一根新的共价键。由于碳原子上原本就缺 电子,新键的一对电子实际都来自于亲核试剂。
于此同时,为了维持羰基碳原子四价,新键形成的同时势必会断开旧键。这里断开的当然是相对而言键能较低的C=O间的π键,异裂后一对电子 转移到氧上,我们将得到阶段性产物:氧负离子中间体。
而这种氧负离子中间体不算稳定,一般会迅速从溶剂中夺取质子,转变为羟基,于是我们得到了亲核加成的最终产物。
如果不管中间过程,只看反应物与最终的生成物,整个反应看起来是C=O间双键变成了单键,C、O原子上各自加上了一个基团或原子。 加上新基团时又存在明显的规律:
原本缺电子的羰基碳与富电子的亲核试剂相连;
而原本富电子的羰基氧与缺电子的质子相连,形成羟基。
这也是整个这一系列亲核加成反应生成物结构上最典型的特征。
上边的历程中,亲核试剂是进攻能力较强的负离子。但有些亲核加成反应中,亲核试剂是电中性的分子(如醇、胺等),亲核能力稍差些。 这时我们往往需要向反应体系中加少量酸作为催化剂,反应历程也会略微发生一些改变,如下图所示。
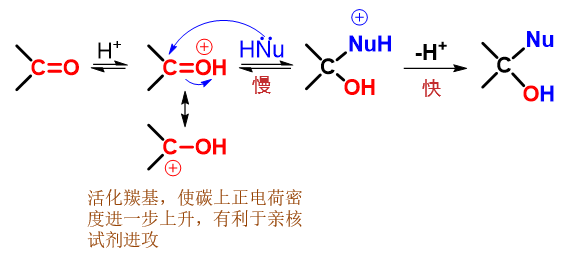
醛酮亲核加成典型历程B:亲核试剂进攻质子化后的羰基
首先,在酸性环境中,羰基里富电子的氧会发生质子化,形成类似[钅羊]盐的结构。与醇或者醚的[钅羊]盐相仿,形成这种结构后也会 使得碳氧间共价键的极性进一步增强,使得碳原子上正电荷密度上升(参见下方的共振极限式),更有利于亲核试剂的进攻。这步质子化 相当于是进一步活化了羰基。
而后,亲核能力不太强的电中性试剂进攻活化了的羰基,引发与之前大同小异的亲核加成。最终脱除质子,得到结构如出一辙的电中性产品。
无论是哪种历程,质子化或脱质子的过程都是比较迅速的,而亲核进攻一步,涉及新键形成旧键断裂,无疑要困难得多,因此总是 反应的速控步。各种亲核加成反应的特性,也就与这一步的关系最为密切。
醛酮的反应活性
反应的速控步,总是富电子的亲核试剂进攻缺电子的羰基碳。在亲核试剂相同的情况下,反应速率基本受到底物醛酮分子两方面的影响:
羰基碳原子上电子云密度(或者说缺电子程度):电子云密度越低,正电荷密度越高,越容易吸引亲核试剂进攻,反应速率也就会越快;
羰基碳旁侧烃基位阻:位阻越小,亲核试剂进攻越容易,反应速率也会越高。
依据这两个原则,我们不难判定不同的羰基化合物反应的相对活性。
比如,对于醛和酮这两种分子,我们不难发现,整体上醛的亲核反应活性会较酮来得高。原因很明显:就位阻而言,酮羰基两侧都是烃基,而醛 羰基一侧是氢原子,位阻上醛比酮要小;就羰基碳电子云密度而言,烃基一般都具有给电子诱导效应,酮羰基两侧两个烃基给电子,醛羰基只有一侧, 因而醛羰基碳的电子云密度也会较酮羰基低一些。两种因素综合,同样条件下醛的反应速率通常要明显高过酮。 而在通常结构的酮当中,羰基一侧是甲基的所谓甲基酮,反应活性通常要高过其它结构的酮——这还是由于位阻的关系。至于普通醛里,活性最高的自然是甲醛了。
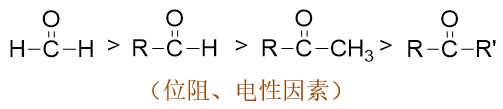
在实验室中我们还经常能遇到羰基旁侧连接芳基的所谓芳香醛酮。相对于脂肪族醛酮而言,芳香醛酮的亲核反应活性一般要差一些。这主要由于电性 方面的缘故。芳香醛酮中羰基与芳基可以形成共轭体系,羰基整体又是个挺强的吸电子基团(-I/-C),尤其是吸电子共轭效应,会把芳基上 的电子往自己身边拉,导致羰基碳上电子云密度上升,正电荷密度下降,亲核反应活性也就随之降低。
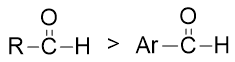
如果芳香醛酮的芳环上还连接有其它基团,则其它基团的电性效应也会对羰基电子云密度造成影响,进而影响亲核反应的活性。下图即为一些典型示例。
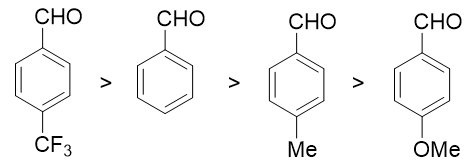
总之,无论何种亲核加成,醛酮的反应活性规律都是类似的。
典型的亲核加成反应
目前我们学习到的较典型与醛酮发生亲核加成反应的试剂,大致有如下几类:
氢氰酸
饱和亚硫酸氢钠水溶液
格氏试剂、烃基锂等金属有机化合物
醇
氨及各类氨衍生物
各类试剂与羰基化合物发生反应的过程大同小异,当然不同的试剂亲核活性存在一定的差异。其中醇、氨及其衍生物, 都是以电中性分子的形式发动亲核进攻,亲核能力相对较弱,因而反应时一般需加入额外的酸作为催化剂以活化羰基(即前述第二种历程)。 氢氰酸与亚硫酸氢钠,进攻试剂是CN-与HSO3-这样的负离子,但由于通常反应条件下溶液中进攻试剂负离子浓度有限, 因而整体亲核能力也不算特别强,只有活性较高的羰基化合物才能与之反应。至于金属有机化合物,亲核能力要强得多, 与各种结构不同的羰基化合物反应一般都没太大的问题。
以下列出各类亲核试剂与醛酮反应时的一些基本要素,供大家复习时参考。
与氢氰酸加成
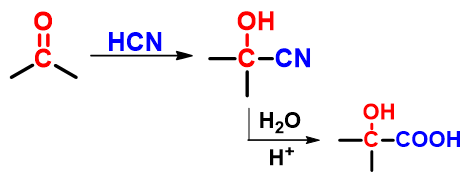
反应
历程
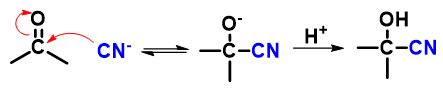
亲核试剂:CN-
反应条件:弱碱性最佳(促进HCN解离出CN-)
反应范围:醛、脂肪族甲基酮、八碳以下环酮,芳香酮无法反应
用途:制备α-羟基酸(亲核加成后再利用腈水解反应)
与饱和亚硫酸氢钠水溶液加成
反应
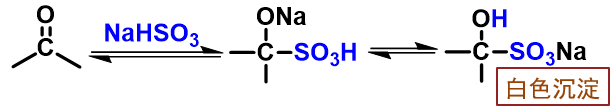
历程
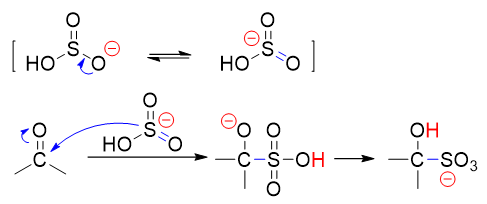
亲核试剂:HSO3-(以S为进攻中心,如上面历程所示,亚硫酸氢根负离子存在一对共振极限式,O/S上均带部分负电荷)
反应条件:饱和亚硫酸氢钠水溶液(不饱和的话亲核试剂浓度不足,一般不易反应)
反应范围:醛、脂肪族甲基酮、八碳以下环酮,芳香酮无法反应(同HCN亲核加成反应基本相同)
格氏试剂等金属有机化合物
反应
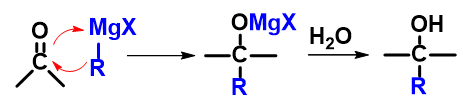
亲核试剂:金属有机化合物中带部分负电荷的烃基部分
反应范围:进攻试剂亲核能力强,几乎所有羰基化合物皆可反应,个别位阻较大者略困难
用途:制备醇的同时延长碳链
备注
使用格氏试剂或其它金属有机化合物从醛酮制备醇的方法,可参加醇酚醚一章的 重难点小结:使用格氏试剂制备醇。
醇
反应
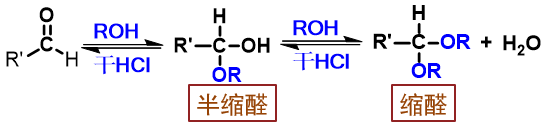
历程
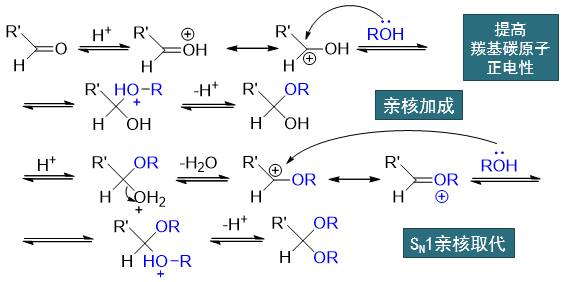
从醛酮得到缩醛、缩酮的过程分两大步:首先得到半缩醛酮的过程,是亲核加成。当然由于醇这种电中性分子亲和能力较差,反应时需要质子性环境,事先活化羰基。 而从半缩醛酮到缩醛酮,实际是个SN1亲核取代,同样也需要质子性环境帮助离去基团羟基的离去。
反应条件:无水强酸催化(常使用干燥HCl)
用途:合成中保护羰基
氨及氨衍生物
氨与单取代氨的反应
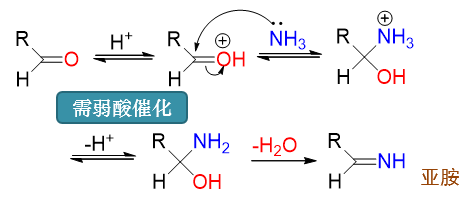
由于电中性氨或氨衍生物分子亲核能力亦不强,这里也需要酸活化羰基。当然氨具有碱性,这里不能用强酸,一般需要使用一些弱酸。 在亲核加成生成α-氨基醇后,由于同一碳上连接了羟基与氨基两个相对活泼的基团,结构不稳定,羟基与氮上的氢会自发消除,得到 C=N双键。
单取代氨与醛酮的反应过程亦类似,同样首先得到α-氨基醇再接脱水,得到C=N双键。
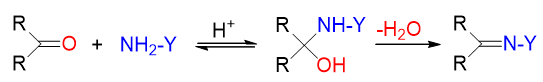
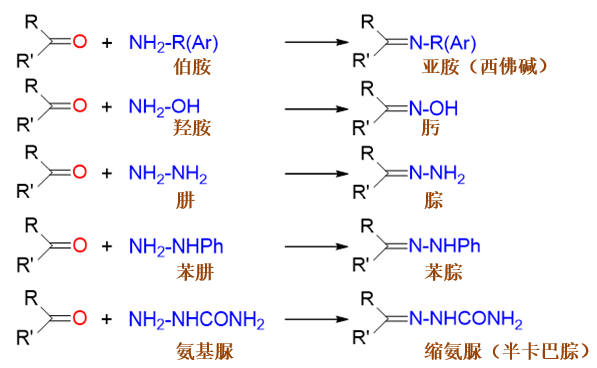
不同的单取代氨衍生物与醛酮反应后,生成结构类似但分类不同的分子。需要特别留神氨衍生物及反应生成物各自的名称:
二取代氨的反应
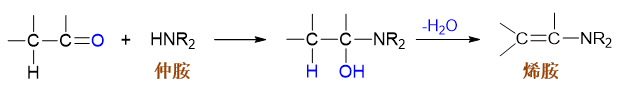
二取代氨(如两个烃基取代氢的仲胺)与醛酮的反应同样是亲核取代,但生成α-氨基醇后,氮原子上不再连接有氢原子,因此羟基只能 转而与原羰基α-碳上的氢消去,生成包含C=C双键的烯胺。
与亲核加成有关的反应
部分醛酮其它类别的反应,其实也与亲核加成有关。典型的如羟醛缩合与Wittig反应。
羟醛缩合
羟醛缩合,我们一般将其归类为醛酮α-H的反应。在通常的碱催化条件下,反应的起点是醛酮酸性α-H的离去,碳负离子的形成。但在此之后, 就是彻头彻尾的亲核加成过程,碳负离子作为亲核试剂,进攻另一分子醛酮的羰基碳,羰基C=O双键转变为单键:
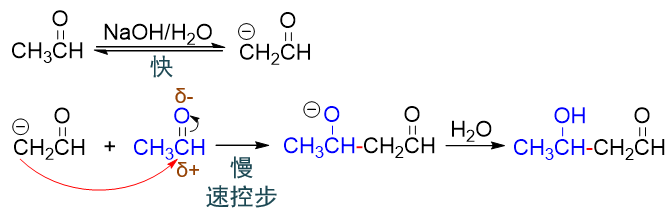
脱α-H形成碳负离子,相对比较容易,整套反应的速控步,还是亲核试剂碳负离子对羰基的进攻。因此羟醛缩合反应的很多特征也与其它的亲核加成 一般无二。
比如产物结构上,与其它亲核加成类似,也是底物分子的羰基转变成羟基,氢加到氧上。而作为亲核试剂的碳负离子,加到底物的羰基碳上。
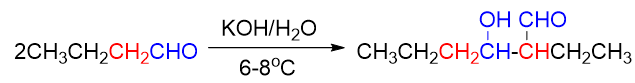
再如反应活性上,也还是醛明显高于酮。醛的反应往往在低温或常温就可以进行,而酮,通常需要加热反应。当然加热时我们也知道,缩合产物 β-羟基醛酮将会发生脱水,得到α,β-不饱和醛酮。
Wittig反应
至于涉及磷叶立德的Wittig反应,从本质上说其实也是一个亲核加成。
我们知道磷叶立德的实际结构是下图中两个共振极限式的平均化,C-P键的成键情况介于单双键之间,而成键电子偏向于碳原子,碳上带有部分负电荷:
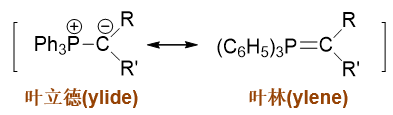
而这种电子云密度较高的碳通常都具有较强的亲核性,它可以进攻羰基中的缺电子碳,引发亲核加成,形成新C-C键的同时,断开羰基C=O间π键,形成 氧负离子:
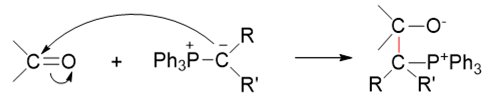
到这里,都与常规的亲核加成反应一般无二。但接下来的过程略有不同。正常亲核加成反应时,生成的氧负离子将从溶剂中夺取一个质子,转变为电中性的羟基。 而对于Wittig反应而言,我们瞅一眼生成的这个中间产物,氧负离子想转变成电中性,压根儿不需要再去找质子,它的旁侧,刚好有个磷正离子。 于是这两者之间迅速配对,形成新的O-P键,关成四元环:
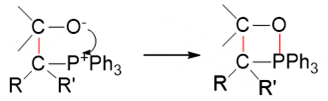
四元环本身张力较大,更何况当前环上还有O、P两个杂原子,很不稳定。几乎在形成的同时,就开始断键破裂,C-O、C-P间共价键发生异裂断开,两对 电子分别转移到O-P、C-C之间,形成O=P、C=C双键,最终,我们生成稳定的产物:三苯氧磷与烯烃。
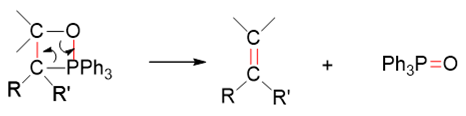
整个Wittig反应的过程连缀起来,大致如下图所示:
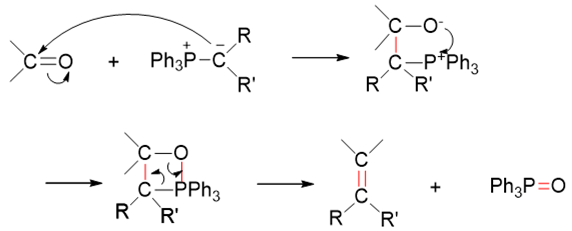
这个过程某种意义上说,我们可以把它看成是一个变形的亲核加成反应,无非是正常的亲核进攻之后,又接了成四元环及四元环破裂的变化。反应的速控步, 还是磷叶立德对羰基亲核进攻的一步,因此反应的很多特征,包括反应活性规律,也与普通的亲核加成一致。
当然,Wittig反应的整个流程看起来比较复杂,在基础有机中并不要求大家掌握。如果不管中间过程,只看头尾的反应物与生成物,其实特别简单: 无非就是醛酮的羰基碳与磷叶立德中的烃基部分,通过新形成的一根C=C双键连接在一起,如是而已:
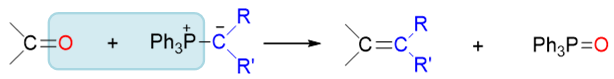
综上,羰基的亲核加成可以说涉及醛酮的最重要的一类化学性质,大量的具体反应都与之相关。我们在学习时,也可以对这些反应进行关联性的记忆。