α,β-不饱和醛酮的亲核加成
机理
普通的醛酮的亲核加成反应,我们已经都很熟悉,反应的动力是富电子亲核试剂对羰基缺电子碳的进攻,进攻的位点非常确定:
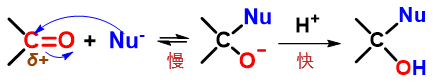
醛酮亲核加成典型历程:亲核试剂直接进攻羰基碳
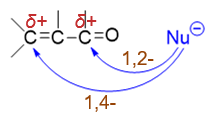
但对于结构特殊、分子内部存在共轭体系的α,β-不饱和醛酮而言,情况会略微发生一些变化。由C=C双键与羰基构成的共轭体系 中,会有两个原子带有部分正电荷,都可能成为亲核试剂进攻的靶点,分别引发1,2-与1,4-加成:
之所以如此,还是与共轭体系与电子离域有关。对于α,β-不饱和醛酮中的电子离域体系,我们可以写出下图中的一系列共振极限式。 这些共振式的写法我们也应当并不陌生,无非就是人为地令电子发生转移。如在经典结构式(I)的基础上,人为令羰基C=O双键中π键 一对电子转移至氧上(箭头a),可以得到电荷分离的共振式(II);而(II)中C2-C4这一区域,我们放眼看过去 其实类似于熟悉的烯丙基碳正离子结构,因而接下来可以再令C3-C4间π键一对电子转移至C2-C3之间形成双键(箭头b),同时电荷也发生转移, 得到共振式(III):
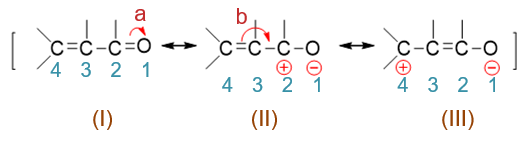
而共振式(II)与(III)中分别是C2、C4带有正电荷,最终分子实际结构是这一系列共振极限式的平均化,2-、3-位碳原子上都会 带有部分正电荷,于是它们皆可能成为亲核试剂进攻的靶点:
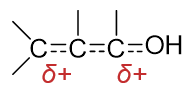
备注
关于共振论的讨论与共振式的写法,若有遗忘,请参见重难点小结:共振论(1)与重难点小结:共振论(1)。
如若亲核试剂进攻2-位碳原子,反应相当于直接发生在羰基上,如下图所示,其过程与普通醛酮的亲核加成也并无二致, 最终产品结构相当于亲核端加在缺电子的2-位碳上,而质子加在富电子的1-位氧上。整个过程我们自然称之为1,2-加成:
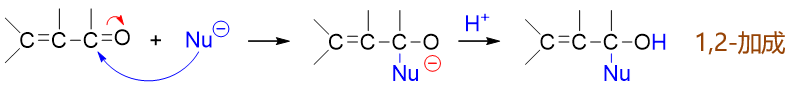
反过来如若亲核试剂进攻4-位碳原子,其过程大致可理解为下图。首先,亲核试剂与C4之间成键,为了保持C4四价,必须 断开键能较低的C3-C4间π键,一对电子转移至C3,形成碳负离子。而这个离子中明显碳负中心与右侧羰基间存在共轭,且 氧的电负性又高于碳,因而我们又可以人为地令C3上多出的一对电子转移至C2-C3之间,形成C=C双键,同时拆开C2-O1之间 的π键,一对电子转移到氧上,得到新的,也是更稳定的共振极限式。而后,质子与负电荷密度更高的氧原子结合,生成电中性 加成产物。就结果来看,亲核端与质子分别加在共轭体系两端的4-、1-位原子上,因此这个过程我们称之为1,4-加成。
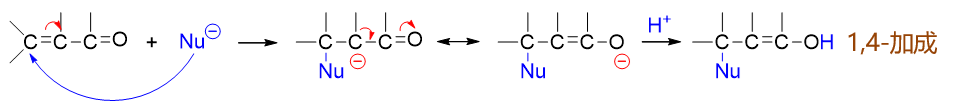
值得注意的是,1,4-加成的结果明显是个烯醇,而我们也知道,烯醇式与酮式之间存在互变异构,且通常酮式来得更加稳定,因此 我们再继续向下写,互变异构后1-位氧上的H又重排到3-位碳上,得到反应的最终产品:
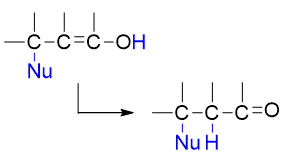
互变异构后的最终产物里,看起来H与Nu分别处于原本共轭体系的3-、4-位,似乎加成就直接发生在C=C双键上,而与羰基无关。 但究其本质,反应实际与共轭体系中所有原子都有关联,先1,4-加成再接互变异构的这么一个过程。
反应倾向
既然α,β-不饱和醛酮的亲核加成存在1,2-与1,4-两条可能的途径,那究竟反应时以何种产品为主?这也是个值得深究的问题。 与之前共轭二烯的亲电共轭加成类似,此处醛酮的亲核共轭加成中,动力学控制容易引发1,2-,而热力学控制容易导致1,4-。 换而言之,反应物(亲核试剂、底物醛酮)活性较高、浓度较高,则倾向1,2-加成;反之则倾向1,4-加成。
如对底物醛酮分子,我们知道醛的亲核加成反应活性要高过酮,因此α,β-不饱和醛相对容易发生1,2-加成,不饱和酮则偏向于 发生1,4-加成。
对于亲核试剂,情况也是类似,活性越高越有利于1,2-,越低越有利于1,4-。常见的亲核试剂与不饱和醛酮发生反应的粗略情况 大致如下表所示:
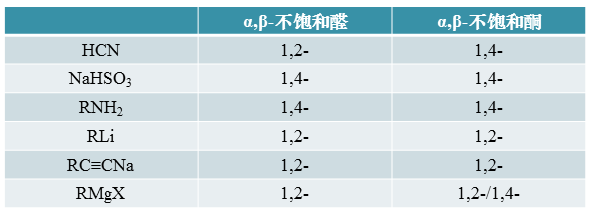
我们明显可以发现,亲核试剂里呈电中性且亲核能力相对较弱的胺(包括其它一些氨衍生物类试剂),一般容易发生1,4-加成。 类似的,活性同样不太高的饱和亚硫酸氢钠水溶液,往往主要发生的也是1,4-加成。反之活性强得多的金属有机化合物, 尤其是烃基锂及炔钠,通常只会导致1,2-加成。而格氏试剂的活性较烃基锂稍弱一些,在遇到羰基旁侧位阻较大的不饱和酮时, 则有可能转而主要进行1,4-加成。
备注
上表仅反映共轭亲核加成的大致情况,实际反应走向往往会更加复杂,还与反应溶剂、温度、时间等多种其它因素等有关联。
最后列举一些典型的反应实例。
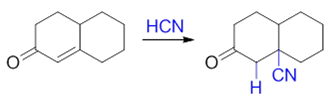
HCN参与反应时,亲核试剂是解离出的浓度不高的氰根负离子,亲核活性不算强,在与不饱和酮的反应中主要进行1,4-加成
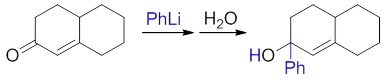
烃基锂试剂亲核活性很强,无论是不饱和醛还是酮,通常主要都是1,2-加成
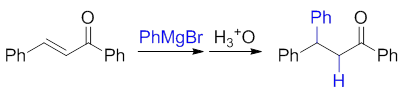
格氏试剂活性稍弱于烃基锂,遇到羰基旁侧位阻较大的不饱和酮,往往主要发生1,4-加成
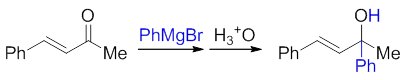
但若羰基旁位阻不大,格氏试剂又会转而以1,2-加成为主
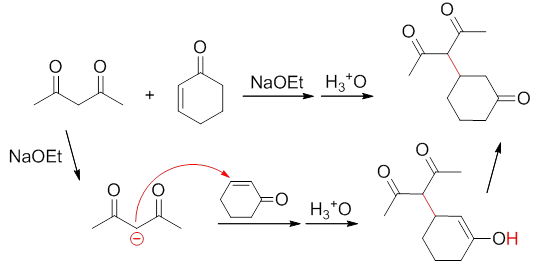
1,3-官能团化合物解离活泼氢得到的碳负离子也具有明显的亲核活性,但解离出的碳负离子浓度一般不会很高,以1,4-加成为主
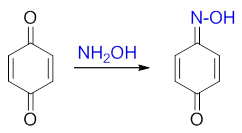
正常氨衍生物与不饱和酮主要发生1,4-加成,但醌活性比普通的不饱和酮更强,较为特殊,与羟胺等反应时常以1,2-加成为主